|
To accurately estimate ligand-receptor binding affinity, quantum chemical optimization and analysis is required to generate "force field consistent" parameters that represent the unique chemistry inherent to each ligand of interest.
Using ab-initio calculations and parameters from known chemistry, each function is derived to provide the most accurate force field parameters possible.
Only then can alchemical methods be used to estimate ligand-receptor binding affinity. |

|
Our technology fully automates this complex and often difficult process.
Ligand → Force Field Parameters → Binding Affinity Estimation
↓
|
The AMOEBA Force field
Sampling issues aside, the most significant errors in the calculation of ligand-receptor binding energies arise from the treatment of electrostatics. Most well known force fields still utilize point charge representations. To improve upon these shortcomings, we utilize the AMOEBA force field, developed by Prof. Jay Ponder at Washington University in St. Louis. This force field computes electrostatic energies far more rigorously, utilizing the following methods: |
|
- The use of multipole expansions to describe the charge distribution surrounding the molecule.
- The effective treatment of charge polarization.
Though these functions add considerable calculation time, rigorous testing shows that they provide a significant boost in accuracy.
↓
|
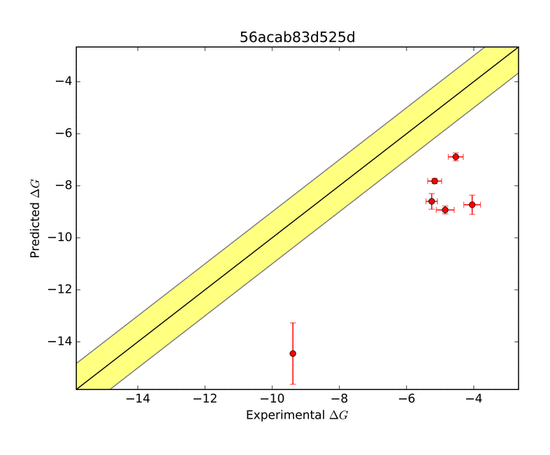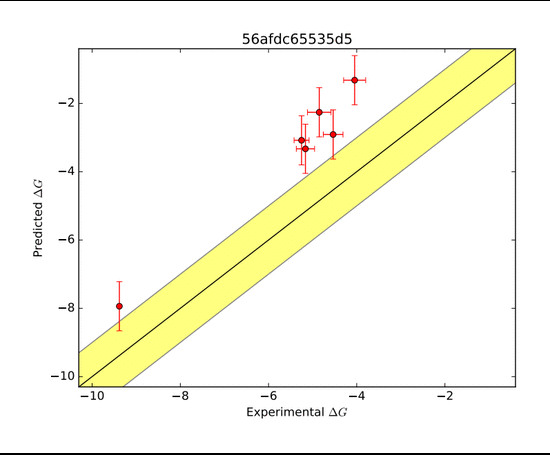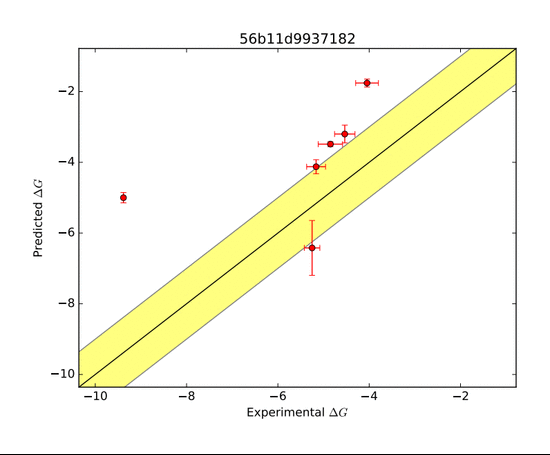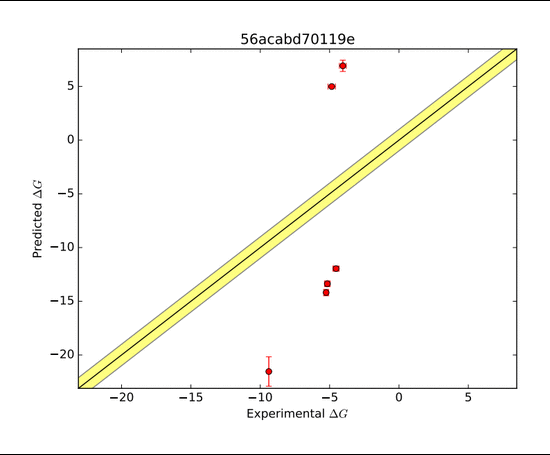
SAMPL is an open blind challenge that enables researchers to test computational methods related to rational drug design, such as the estimation of binding affinity and hydration free energy. Shown here was one such host-guest complex (CB Clip) with numerous diverse ligands that was presented to the competitors in SAMPL5. The goal was to accurately predict the ΔG of binding.
|

|
The AMOEBA force field performed very well in predicting the ΔG of binding. The green zone indicates a prediction within 1 kcal/mol of the target, and is considered very good. The other groups who submitted entries for this system were far less successful as shown in the series of predictions below.

|
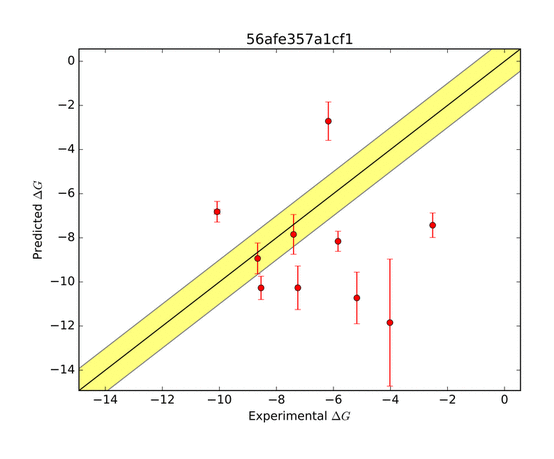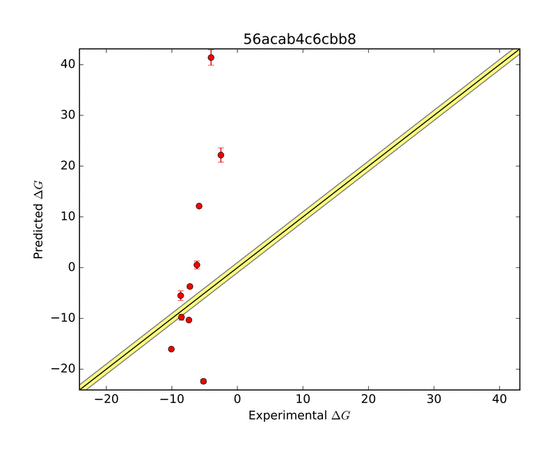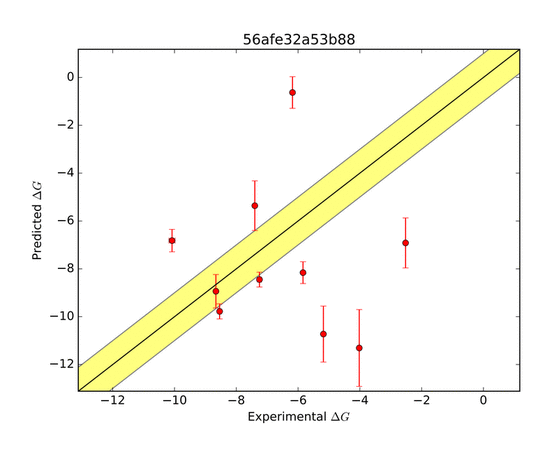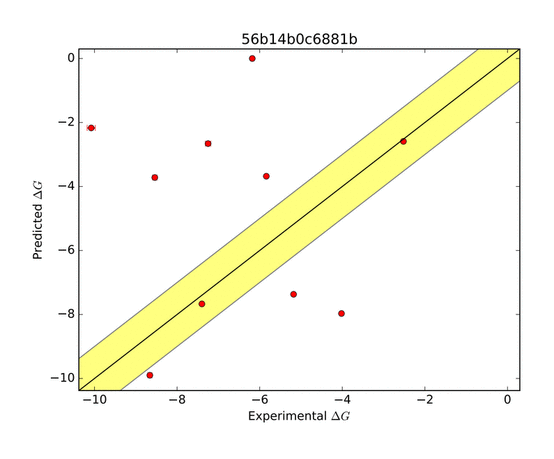
|
↓
|
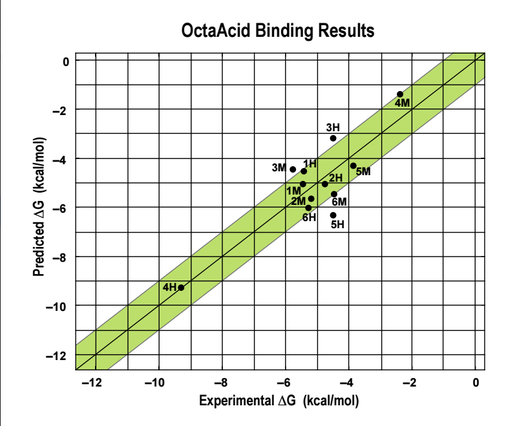
Shown here was another host-guest complex (Octa Acid) with several diverse ligands that was presented to the competitors in SAMPL5. The goal was again to accurately predict the ΔG of binding. In this host, the R groups could be either hydrogen (H) or methyl groups (M).
The AMOEBA force field again performed very well in predicting the ΔG of binding. The AMOEBA graph has both (R=H) and (R=Me) predictions combined, while the other group's data are in separate graphs. Although several groups were able to represent trends in a general sense, they were not as accurate throughout the range of compounds as AMOEBA was.
|

|
|
|